जीनोमिक्स
जीनोमिक्स
जीनोमिक्स जीव विज्ञान का एक अंतःविषय क्षेत्र है, जो जीनोम की संरचना, कार्य, विकास, मानचित्रण और संपादन पर केंद्रित है। एक जीनोम एक जीव का डीएनए का पूरा सेट है, जिसमें उसके सभी जीन शामिल हैं। आनुवांशिकी के विपरीत, जो व्यक्ति जीन के अध्ययन और वंशानुक्रम में उनकी भूमिका को संदर्भित करता है, जीनोमिक्स का उद्देश्य किसी जीव के जीन के सभी के सामूहिक लक्षण वर्णन और परिमाणीकरण, उनके अंतर्संबंध हैं। और जीव पर प्रभाव। जीन एंजाइम और मैसेंजर अणुओं की सहायता से प्रोटीन के उत्पादन को निर्देशित कर सकते हैं। बदले में, प्रोटीन शरीर की संरचनाओं जैसे अंगों और ऊतकों और साथ ही रासायनिक प्रतिक्रियाओं को नियंत्रित करते हैं और कोशिकाओं के बीच संकेतों को ले जाते हैं। जीनोमिक्स में उच्च थ्रूपुट डीएनए अनुक्रमण और जैव सूचना विज्ञान के उपयोग के माध्यम से जीनोम की अनुक्रमण और विश्लेषण को शामिल करना और पूरे जीनोम के कार्य और संरचना का विश्लेषण करना शामिल है। जीनोमिक्स में अग्रिमों ने खोज-आधारित अनुसंधान और सिस्टम बायोलॉजी में एक क्रांति ला दी है, यहां तक कि मस्तिष्क जैसे सबसे जटिल जैविक प्रणालियों की समझ को सुविधाजनक बनाने के लिए।
क्षेत्र में इंट्रोजेनोमिक (जीनोम के भीतर) की घटनाओं का अध्ययन भी शामिल है। जैसे कि एपिस्टासिस (दूसरे पर एक जीन का प्रभाव), प्लियोट्रॉपी (एक जीन जो एक से अधिक लक्षणों को प्रभावित करता है), हेटेरोसिस (हाइब्रिड वाइगर) और जीनोम के भीतर लोकी और एलील्स के बीच अन्य इंटरैक्शन।
सामग्रीइतिहास
व्युत्पत्ति
ग्रीक से From जीन , "जीन" (गामा, एप्सिलॉन, एनयू, एप्सिलॉन) का अर्थ "बन" जाता है। बनाएँ, निर्माण, जन्म ", और बाद के संस्करण: वंशावली, उत्पत्ति, आनुवांशिकी, जीनिक, जीनोमेरे, जीनोटाइप, जीनस आदि। शब्द जीनोम (जर्मन जीनोम से, हेंस विंकलर को जिम्मेदार ठहराया गया) 1926 की शुरुआत में अंग्रेजी में प्रयोग किया गया था, इस शब्द जीनोमिक्स को एक बैठक में बीयर ओवर में जैक्सन लेबोरेटरी (बार हार्बर, मेन) के एक आनुवंशिकीविद टॉम रॉडरिक द्वारा गढ़ा गया था। मैं n 1986 में मानव जीनोम की मैपिंग पर मैरीलैंड।
प्रारंभिक अनुक्रमण के प्रयास
डीएनए के पेचदार संरचना की पुष्टि रोसालिंड फ्रेंकलिन के बाद, जेम्स डी। वाटसन और फ्रांसिस क्रिक का प्रकाशन 1953 में डीएनए की संरचना और 1955 में इंसुलिन के एमिनो एसिड अनुक्रम के फ्रेड सेंगर के प्रकाशन, न्यूक्लिक एसिड अनुक्रमण प्रारंभिक आणविक जीवविज्ञानी का एक प्रमुख लक्ष्य बन गया। 1964 में, रॉबर्ट डब्ल्यू। होली और सहकर्मियों ने पहले न्यूक्लिक एसिड अनुक्रम को निर्धारित किया, जो कि एलेन ट्रांसफर आरएनए के राइबोन्यूक्लियोटाइड अनुक्रम था। इस काम को आगे बढ़ाते हुए, मार्शल निरेनबर्ग और फिलिप लेडर ने आनुवंशिक कोड की ट्रिपल प्रकृति का पता लगाया और अपने प्रयोगों में 64 में से 54 कोडन के अनुक्रमों को निर्धारित करने में सक्षम थे। 1972 में, यूनिवर्सिटी ऑफ़ गेंट (गेन्ट, बेल्जियम) के मॉलिक्यूलर बायोलॉजी की प्रयोगशाला में वाल्टर फ़िएर्स और उनकी टीम ने जीन के क्रम को निर्धारित करने वाले पहले व्यक्ति थे: जीवाणु के लिए जीन MS2 कोट प्रोटीन। फीयर के समूह ने अपने MS2 कोट प्रोटीन कार्य पर विस्तार किया, जिससे जीवाणुनाशक MS2-RNA (जिसका जीनोम केवल चार जीन 3569 बेस पेयर में रहता है) और सिमीयन वायरस 40, क्रमशः 1976 और 1978 में पूरा होने का निर्धारण करते हैं।
डीएनए-अनुक्रमण तकनीक विकसित हुई
इंसुलिन के अमीनो एसिड अनुक्रम पर अपने सेमिनल काम के अलावा, फ्रेडरिक सेंगर और उनके सहयोगियों ने डीएनए अनुक्रमण तकनीक के विकास में महत्वपूर्ण भूमिका निभाई जिसने व्यापक की स्थापना को सक्षम किया जीनोम अनुक्रमण परियोजनाओं। 1975 में, उन्होंने और एलन कॉल्सन ने रेडियोलिंबेड न्यूक्लियोटाइड्स के साथ डीएनए पोलीमरेज़ का उपयोग करके एक अनुक्रमण प्रक्रिया प्रकाशित की जिसे उन्होंने प्लस और माइनस तकनीक कहा। इसमें दो निकट संबंधी तरीके शामिल थे जो परिभाषित 3 'टर्मिनी के साथ लघु ओलिगोन्यूक्लियोटाइड उत्पन्न करते थे। ये एक polyacrylamide जेल (polyacrylamide जेल वैद्युतकणसंचलन कहा जाता है) पर वैद्युतकणसंचलन द्वारा विभाजित किया जा सकता है और ऑटोरैडियोग्राफी का उपयोग कर कल्पना की। यह प्रक्रिया एक बार में 80 न्यूक्लियोटाइड तक अनुक्रम कर सकती है और एक बड़ा सुधार था, लेकिन अभी भी बहुत श्रमसाध्य था। फिर भी, 1977 में उनका समूह एकल-फंसे हुए बैक्टीरियोफेज haX174 के 5,386 न्यूक्लियोटाइड्स में से अधिकांश का अनुक्रम करने में सक्षम था, जो पहले पूरी तरह से अनुक्रमित डीएनए-आधारित जीनोम को पूरा करता था। प्लस और माइनस के शोधन के परिणामस्वरूप श्रृंखला-समाप्ति, या सेंगर विधि (नीचे देखें) हुई, जिसने डीएनए अनुक्रमण, जीनोम मैपिंग, डेटा भंडारण, और जैव सूचनात्मक विश्लेषण की तकनीकों का आधार बनाया। सबसे व्यापक रूप से अनुसंधान के निम्नलिखित तिमाही-शताब्दी में उपयोग किया जाता है। उसी वर्ष हार्वर्ड विश्वविद्यालय के वाल्टर गिल्बर्ट और एलन मैक्सम ने डीएनए अनुक्रमण के मैक्सम-गिल्बर्ट विधि (जिसे रासायनिक विधि के रूप में भी जाना जाता है) को विकसित किया, जिसमें ज्ञात आधारों पर डीएनए के तरजीही दरार शामिल हैं, एक कम भी। कुशल विधि। न्यूक्लिक एसिड की अनुक्रमण में उनके जमीनी स्तर पर काम के लिए, गिलबर्ट और सेंगर ने पॉल बर्ग (पुनः संयोजक डीएनए) के साथ रसायन विज्ञान में आधा 1980 का नोबेल पुरस्कार साझा किया।
पूरा जीनोम
।इन प्रौद्योगिकियों के आगमन से जीनोम अनुक्रमण परियोजनाओं के पूरा होने की गुंजाइश और गति में तेजी से सुधार हुआ। यूकेरियोटिक ऑर्गेनेल का पहला पूर्ण जीनोम अनुक्रम, मानव माइटोकॉन्ड्रियन (16,568 बीपी, लगभग 16.6 केबी), 1981 में रिपोर्ट किया गया था और 1986 में इसके बाद पहला क्लोरोप्लास्ट जीनोम। 1992 में, शराब बनाने वाले खमीर का पहला यूकेरियोटिक गुणसूत्र, गुणसूत्र III <। i> Saccharomyces cerevisiae (315 kb) का अनुक्रम किया गया। 1995 में पहली बार जीवित रहने वाले जीव का हीमोफिलस इन्फ्लुएंजा (1.8 एमबी) था। अगले वर्ष उत्तरी अमेरिका, यूरोप और जापान के प्रयोगशालाओं के शोधकर्ताओं का एक संघ पूरा होने की घोषणा की। एक यूकेरियोट का पहला पूर्ण जीनोम अनुक्रम, एस। cerevisiae (12.1 Mb), और तब से जीनोमों ने तेजी से बढ़ती गति के साथ अनुक्रम जारी रखा है। अक्टूबर 2011 तक, पूर्ण अनुक्रम उपलब्ध हैं: 2,719 वायरस, 1,115 आर्किया और बैक्टीरिया, और 36 यूकेरियोट्स, जिनमें से लगभग आधे कवक हैं।
अधिकांश सूक्ष्मजीव जिनके जीनोम पूरी तरह से अनुक्रमित हैं, समस्याग्रस्त हैं। रोगजनकों, जैसे कि हीमोफिलस इन्फ्लुएंजा , जिसके परिणामस्वरूप माइक्रोबियल विविधता की चौड़ाई की तुलना में उनके फाइटोलैनेटिक वितरण में एक स्पष्ट पूर्वाग्रह है। अन्य अनुक्रमित प्रजातियों में से, अधिकांश को चुना गया क्योंकि वे अच्छी तरह से अध्ययन किए गए मॉडल जीव थे या अच्छे मॉडल बनने का वादा करते थे। खमीर ( Saccharomyces cerevisiae ) लंबे समय से यूकेरियोटिक कोशिका के लिए एक महत्वपूर्ण मॉडल जीव है, जबकि फल मक्खी ड्रोसोफिला मेलानोगास्टर एक बहुत महत्वपूर्ण उपकरण रहा है (विशेष रूप से प्रारंभिक प्री-आणविक में) जेनेटिक्स)। कृमि सेनोरहाडाइटिस एलिगेंस बहुकोशिकीय जीवों के लिए अक्सर इस्तेमाल किया जाने वाला सरल मॉडल है। Zebrafish Brachydanio rerio का उपयोग आणविक स्तर पर कई विकासात्मक अध्ययनों के लिए किया जाता है, और संयंत्र Arabidopsis thaliana फूलों के पौधों के लिए एक मॉडल जीव है। जापानी पफ़रफ़िश ( तकिफ़ुगु रुब्रिज़ ) और धब्बेदार हरे रंग का पफ़रफ़िश ( टेट्रोडॉन निग्रोविरिडिस ) अपने छोटे और कॉम्पैक्ट जीनोम के कारण दिलचस्प हैं, जिनमें अधिकांश प्रजातियों की तुलना में बहुत कम नॉनकोडिंग डीएनए होते हैं। । स्तनधारी कुत्ता ( कैनिस परिचित ), भूरे रंग का चूहा ( रेटस नॉरवेगिकस ), माउस ( मूस मस्कुलस ), और चिंपांज़ी ( पान ट्रोग्लोडाइट्स) चिकित्सा अनुसंधान में सभी महत्वपूर्ण मॉडल पशु हैं
मानव जीनोम के किसी न किसी मसौदे को ह्यूमन जीनोम प्रोजेक्ट ने 2001 की शुरुआत में पूरा किया, जिससे बहुत अधिक धूमधाम पैदा हुई। 2003 में पूरी हुई इस परियोजना ने एक विशिष्ट व्यक्ति के लिए पूरे जीनोम को अनुक्रमित किया, और 2007 तक इस अनुक्रम को "समाप्त" (20,000 ठिकानों में एक त्रुटि से कम और सभी गुणसूत्र इकट्ठे हुए) घोषित किया गया। उसके बाद के वर्षों में, कई अन्य व्यक्तियों के जीनोम को अनुक्रमित किया गया है, आंशिक रूप से 1000 जीनोम प्रोजेक्ट के तत्वावधान में, जिसने अक्टूबर 2012 में 1,092 जीनोम की अनुक्रमण की घोषणा की। इस परियोजना का समापन नाटकीय रूप से अधिक के विकास से संभव हुआ। कुशल अनुक्रमण प्रौद्योगिकियों और एक बड़े अंतरराष्ट्रीय सहयोग से महत्वपूर्ण जैव सूचना विज्ञान संसाधनों की प्रतिबद्धता की आवश्यकता है। मानव जीनोमिक डेटा के निरंतर विश्लेषण ने मानव समाजों के लिए गहरा राजनीतिक और सामाजिक प्रतिक्षेप किया है।
"omics" क्रांति
अंग्रेजी भाषा के नवविज्ञान omics सूचना विज्ञान अध्ययन के एक क्षेत्र को संदर्भित करता है। जीवविज्ञान -omics में समाप्त हो रहा है, जैसे कि जीनोमिक्स, प्रोटिओमिक्स या मेटाबॉलिक। संबंधित प्रत्यय-क्रम का उपयोग ऐसे क्षेत्रों के अध्ययन की वस्तुओं को संबोधित करने के लिए किया जाता है, जैसे कि जीनोम, प्रोटिओम या चयापचय। आणविक जीव विज्ञान में प्रयुक्त प्रत्यय -ome किसी प्रकार के समग्रता को संदर्भित करता है; आम तौर पर ओमेक्स बड़े, व्यापक जैविक डेटा सेटों के अध्ययन का उल्लेख करते हैं। हालांकि इस शब्द के उपयोग में वृद्धि ने कुछ वैज्ञानिकों (जोनाथन ईसेन, दूसरों के बीच) को यह दावा करने के लिए प्रेरित किया है कि यह ओवरसोल्ड हो गया है, यह सभी घटकों के पूर्ण या निकट-पूर्ण वर्गीकरण के मात्रात्मक विश्लेषण की दिशा में परिवर्तन को दर्शाता है। एक प्रणाली। उदाहरण के लिए, सहजीवन के अध्ययन में, शोधकर्ता जो कभी एक एकल जीन उत्पाद के अध्ययन तक सीमित थे, अब एक साथ कई प्रकार के जैविक अणुओं के कुल पूरक की तुलना कर सकते हैं।
जीनोम विश्लेषण
<। p> एक जीव के चुने जाने के बाद, जीनोम परियोजनाओं में तीन घटक शामिल होते हैं: डीएनए का अनुक्रमण, मूल गुणसूत्र का प्रतिनिधित्व बनाने के लिए उस क्रम की असेंबली, और उस प्रतिनिधित्व का एनोटेशन और विश्लेषण।ऐतिहासिक रूप से, अनुक्रमण अनुक्रमण केंद्रों में किया गया था, केंद्रीयकृत सुविधाओं (बड़े स्वतंत्र संस्थानों जैसे कि संयुक्त जीनोम संस्थान से जो एक वर्ष में दर्जनों टेररोकेशनों को स्थानीय आणविक जीव विज्ञान कोर सुविधाओं तक पहुंचाते हैं) जो शोध करते हैं महंगे उपकरण और तकनीकी सहायता के साथ प्रयोगशालाएं आवश्यक हैं। चूंकि अनुक्रमण तकनीक में सुधार जारी है, हालांकि, प्रभावी शैक्षणिक बदलाव की एक नई पीढ़ी औसत शैक्षणिक प्रयोगशाला की पहुंच के भीतर आई है। कुल मिलाकर, जीनोम अनुक्रमण दृष्टिकोण दो व्यापक श्रेणियों में आते हैं, शॉटगन और उच्च-थ्रूपुट (या अगली पीढ़ी ) अनुक्रमण । >
शॉटगन सीक्वेंसिंग एक सीक्वेंसिंग विधि है जो डीएनए के दृश्यों के विश्लेषण के लिए 1000 बेस जोड़े से अधिक, पूरे क्रोमोसोम तक और शामिल है। यह एक बन्दूक के तेजी से विस्तार, अर्ध-यादृच्छिक फायरिंग पैटर्न के साथ सादृश्य द्वारा नामित किया गया है। चूंकि जेल वैद्युतकणसंचलन अनुक्रमण केवल काफी कम अनुक्रमों (100 से 1000 आधार जोड़े) के लिए इस्तेमाल किया जा सकता है, अब डीएनए अनुक्रमों को यादृच्छिक छोटे खंडों में तोड़ा जाना चाहिए जो कि रीड्स प्राप्त करने के लिए अनुक्रमित होते हैं। लक्ष्य डीएनए के लिए कई अतिव्यापी रीडिंग इस विखंडन और अनुक्रमण के कई दौर प्रदर्शन करके प्राप्त की जाती हैं। कंप्यूटर प्रोग्राम तब एक निरंतर अनुक्रम में उन्हें इकट्ठा करने के लिए विभिन्न रीड्स के ओवरलैपिंग सिरों का उपयोग करते हैं। शॉटगन अनुक्रमण एक यादृच्छिक नमूनाकरण प्रक्रिया है, किसी दिए गए न्यूक्लियोटाइड को सुनिश्चित करने के लिए ओवर-सैंपलिंग की आवश्यकता होती है जिसे पुन: निर्मित अनुक्रम में दर्शाया जाता है; रीड की औसत संख्या जिसके द्वारा एक जीनोम को ओवर-सैंपल किया जाता है, उसे कवरेज कहा जाता है।
अपने इतिहास के लिए, शॉटगन अनुक्रमण की अंतर्निहित तकनीक शास्त्रीय श्रृंखला-समाप्ति विधि या 'सेंगर विधि' थी, जो कि इन विट्रो डीएनए प्रतिकृति के दौरान डीएनए पोलीमरेज़ द्वारा चेन-टर्मिनेटिंग डिडॉक्सिन्यूक्लियोटाइड्स के चयनात्मक निगमन पर आधारित है। हाल ही में, उच्च-थ्रूपुट अनुक्रमण विधियों द्वारा शॉटगन अनुक्रमण को विशेष रूप से बड़े पैमाने पर, स्वचालित जीनोम विश्लेषण के लिए दबा दिया गया है। हालांकि, सेंगर विधि व्यापक रूप से उपयोग में रहती है, मुख्य रूप से छोटे पैमाने की परियोजनाओं के लिए और विशेष रूप से लंबे समय तक सन्निहित डीएनए अनुक्रम प्राप्त करने के लिए (& gt; 500 न्यूक्लियोटाइड्स)। चेन-टर्मिनेशन के तरीकों के लिए सिंगल-फंसे हुए डीएनए टेम्प्लेट, एक डीएनए प्राइमर, एक डीएनए पोलीमरेज़, नॉर्मल डीऑक्सीन्यूक्लियोसेट्रिफ़ोफ़ेट्स (dNTPs) और संशोधित न्यूक्लियोटाइड्स (dideoxyNTPs) की ज़रूरत होती है जो डीएनए स्ट्रैंड बढ़ाव को समाप्त करते हैं। इन चेन-टर्मिनेटिंग न्यूक्लियोटाइड्स में दो न्यूक्लियोटाइड्स के बीच फॉस्फोडाइस्टर बॉन्ड के निर्माण के लिए आवश्यक 3'-OH समूह की कमी होती है, जिससे डीएनए पोलीमरेज़ डीएनए के विस्तार को रोक देता है जब एक ddNTP को शामिल किया जाता है। DdNTPs को रेडियो सिक्वेंसर या डीएनए सीक्वेंसर में डिटेक्ट करने के लिए लेबल किया जा सकता है। आमतौर पर, ये मशीनें एक ही बैच (रन) में 96 डीएनए नमूने एक दिन में 48 तक चला सकती हैं।
कम लागत वाली अनुक्रमण की उच्च मांग ने उच्च-थ्रूपुट अनुक्रमण के विकास को प्रेरित किया है। ऐसी तकनीकें जो अनुक्रमण प्रक्रिया को समानांतर बनाती हैं, एक साथ हजारों या लाखों क्रम का निर्माण करती हैं। उच्च-थ्रूपुट अनुक्रमण का उद्देश्य मानक डाई-टर्मिनेटर विधियों के साथ डीएनए अनुक्रमण की लागत को कम करना है। अल्ट्रा-हाई-थ्रूपुट अनुक्रमण में, लगभग 500,000 अनुक्रमण-द्वारा-संश्लेषण प्रक्रियाएं समानांतर में चलाई जा सकती हैं।
इलुमिना डाई अनुक्रमण विधि प्रतिवर्ती डाई-टर्मिनेटरों पर आधारित है और 1996 में इसे विकसित किया गया था। पास्कल मेयर और लॉरेंट फारिनेली द्वारा जिनेवा बायोमेडिकल रिसर्च इंस्टीट्यूट। इस पद्धति में, डीएनए अणुओं और प्राइमरों को पहले एक स्लाइड पर जोड़ा जाता है और पोलीमरेज़ के साथ प्रवर्धित किया जाता है ताकि स्थानीय क्लोनल कॉलोनियों, शुरू में "डीएनए कॉलोनियों" को गढ़ा जाए। अनुक्रम को निर्धारित करने के लिए, चार प्रकार के प्रतिवर्ती टर्मिनेटर बेस (आरटी-बेस) जोड़े जाते हैं और गैर-शामिल न्यूक्लियोटाइड को धोया जाता है। पाइरोडिंग के विपरीत, डीएनए श्रृंखलाओं को एक समय में एक न्यूक्लियोटाइड बढ़ाया जाता है और एक ही समय में छवि अधिग्रहण को देरी से किया जा सकता है, जिससे एक ही कैमरे से ली गई अनुक्रमिक छवियों द्वारा डीएनए कॉलोनियों के बहुत बड़े सरणियों को कैप्चर करने की अनुमति मिलती है। एंजाइमैटिक रिएक्शन और इमेज कैप्चर को घटाना इष्टतम थ्रूपुट और सैद्धांतिक रूप से असीमित अनुक्रमण क्षमता के लिए अनुमति देता है; एक इष्टतम विन्यास के साथ, साधन का अंतिम प्रवाह केवल कैमरे के ए / डी रूपांतरण दर पर निर्भर करता है। कैमरा फ्लोरोसेंटली लेबल न्यूक्लियोटाइड्स की छवियों को लेता है, फिर टर्मिनल 3 'ब्लॉकर के साथ डाई को रासायनिक रूप से डीएनए से हटा दिया जाता है, जिससे अगला चक्र
हो सकता है।एक वैकल्पिक दृष्टिकोण, आयन सेमीकंडक्टर अनुक्रमण, मानक डीएनए प्रतिकृति रसायन विज्ञान पर आधारित है। यह तकनीक एक हाइड्रोजन आयन की रिहाई को मापती है जिसमें हर बार एक बेस को शामिल किया जाता है। टेम्प्लेट डीएनए युक्त एक माइक्रोवेल एक एकल न्यूक्लियोटाइड से भर जाता है, अगर न्यूक्लियोटाइड टेम्पलेट स्ट्रैंड के पूरक है तो इसे शामिल किया जाएगा और एक हाइड्रोजन आयन जारी किया जाएगा। यह रिलीज़ ISFET आयन सेंसर को ट्रिगर करता है। यदि एक होमोपोलिमर टेम्पलेट अनुक्रम में मौजूद है, तो एक ही बाढ़ चक्र में कई न्यूक्लियोटाइड शामिल किए जाएंगे, और पता चला विद्युत संकेत आनुपातिक रूप से अधिक होगा।
विधानसभा
अनुक्रम विधानसभा संरेखण को संदर्भित करता है। और मूल अनुक्रम को फिर से संगठित करने के लिए बहुत लंबे डीएनए अनुक्रम के टुकड़े का विलय। यह वर्तमान डीएनए अनुक्रमण तकनीक के रूप में आवश्यक है, एक निरंतर अनुक्रम के रूप में पूरे जीनोम को नहीं पढ़ सकता है, बल्कि उपयोग की गई तकनीक के आधार पर, 20 से 1000 के बीच के छोटे टुकड़ों को पढ़ता है। तीसरी पीढ़ी की अनुक्रमण तकनीकें जैसे कि PacBio या ऑक्सफ़ोर्ड नैनोपोर नियमित रूप से अनुक्रमण रीडिंग & gt; 10 केबी लंबाई में उत्पन्न करती हैं; हालाँकि, उनकी उच्च त्रुटि दर लगभग 15 प्रतिशत है। आमतौर पर शॉर्ट फ्रैगमेंट, जिसे रीड कहा जाता है, शॉटगन सिक्वेंसिंग जीनोमिक डीएनए या जीन ट्रांसक्रिप्शंस (ईएसटी) के परिणामस्वरूप होता है।
असेंबली को मोटे तौर पर दो दृष्टिकोणों में वर्गीकृत किया जा सकता है: डे नोवो असेंबली, के लिए। जीनोम जो अतीत में किसी भी अनुक्रम के समान नहीं हैं, और तुलनात्मक विधानसभा, जो विधानसभा के दौरान संदर्भ के रूप में निकट संबंधी जीव के मौजूदा अनुक्रम का उपयोग करता है। तुलनात्मक असेंबली के सापेक्ष, डे नोवो असेंबली कम्प्यूटेशनल रूप से कठिन (एनपी-हार्ड) है, जिससे यह शॉर्ट-रीड एनजीएस प्रौद्योगिकियों के लिए कम अनुकूल है। डे नोवो असेंबली प्रतिमान में असेंबली, एयूरियन पथ रणनीतियों और ओवरलैप-लेआउट-सर्वसम्मति (ओएलसी) रणनीतियों के लिए दो प्राथमिक रणनीतियां हैं। OLC रणनीतियाँ अंततः ओवरलैप ग्राफ के माध्यम से हैमिल्टन पथ बनाने की कोशिश करती हैं जो कि NP-कठिन समस्या है। यूलरिअन पथ रणनीतियाँ कम्प्यूटेशनल रूप से अधिक ट्रैक्टेबल हैं क्योंकि वे एक डिबिजिन ग्राफ के माध्यम से एक यूलरियन मार्ग खोजने की कोशिश करते हैं।
समाप्त जीनोम को एक एकल सन्निहित अनुक्रम के रूप में परिभाषित किया गया है जिसमें प्रत्येक प्रतिकृति का प्रतिनिधित्व करने वाले कोई अस्पष्टता नहीं है।
एनोटेशन
अकेले डीएनए अनुक्रम विधानसभा अतिरिक्त विश्लेषण के बिना कम मूल्य की है। जीनोम एनोटेशन अनुक्रमों के लिए जैविक जानकारी संलग्न करने की प्रक्रिया है, और इसमें तीन मुख्य चरण होते हैं:
- जीनोम के उन हिस्सों की पहचान करना जो प्रोटीन के लिए कोड नहीं करते हैं
- तत्वों की पहचान करना जीनोम, एक प्रक्रिया जिसे जीन पूर्वानुमान कहा जाता है, और
- इन तत्वों के लिए जैविक जानकारी संलग्न करता है।
स्वचालित एनोटेशन उपकरण इन चरणों को निष्पादित करने का प्रयास करते हैं सिलिको में /। i>, मैनुअल एनोटेशन (उर्फ क्यूरेशन) के विपरीत, जिसमें मानव विशेषज्ञता और संभावित प्रयोगात्मक सत्यापन शामिल है। आदर्श रूप में, ये सह-अस्तित्व में आते हैं और एक ही एनोटेशन पाइपलाइन में एक दूसरे को पूरक करते हैं (नीचे भी देखें)।
परंपरागत रूप से, एनोटेशन का मूल स्तर समानताएं खोजने के लिए BLAST का उपयोग कर रहा है, और फिर होमलोन के आधार पर जीनोम की व्याख्या करता है। । हाल ही में, एनोटेशन प्लेटफॉर्म पर अतिरिक्त जानकारी जोड़ी गई है। अतिरिक्त जानकारी मैनुअल एनोटेटर्स को जीन के बीच विसंगतियों को कम करने की अनुमति देती है जिन्हें समान एनोटेशन दिया जाता है। कुछ डेटाबेस अपने सबसिस्टम दृष्टिकोण के माध्यम से जीनोम एनोटेशन प्रदान करने के लिए जीनोम संदर्भ जानकारी, समानता स्कोर, प्रयोगात्मक डेटा और अन्य संसाधनों के एकीकरण का उपयोग करते हैं। अन्य डेटाबेस (जैसे एनसेम्बल) दोनों क्यूरेटेड डेटा स्रोतों के साथ-साथ अपने स्वचालित जीनोम एनोटेशन पाइपलाइन में सॉफ्टवेयर टूल्स की एक श्रृंखला पर भरोसा करते हैं। संरचनात्मक एनोटेशन में जीनोमिक तत्वों, मुख्य रूप से ओआरएफ और उनके स्थानीयकरण, या जीन संरचना की पहचान शामिल है। कार्यात्मक एनोटेशन में जीनोमिक तत्वों के लिए जैविक जानकारी संलग्न करना शामिल है।
पाइपलाइनों और डेटाबेस को अनुक्रमित करना
बड़ी मात्रा में डेटा से संबंधित प्रतिलिपि प्रस्तुत करने और कुशल प्रबंधन की आवश्यकता जीनोम परियोजनाओं के साथ अभिप्राय है कि कम्प्यूटेशनल पाइपलाइनों के जीनोमिक्स में महत्वपूर्ण अनुप्रयोग हैं।
अनुसंधान क्षेत्र
कार्यात्मक जीनोमिक्स
कार्यात्मक जीनोमिक्स आणविक जीव विज्ञान का एक क्षेत्र है जो जीन (और प्रोटीन) कार्यों और इंटरैक्शन का वर्णन करने के लिए जीनोमिक परियोजनाओं (जैसे जीनोम अनुक्रमण परियोजनाओं) द्वारा उत्पादित डेटा की विशाल संपत्ति का उपयोग करने का प्रयास करता है। कार्यात्मक जीनोमिक्स जीन प्रतिलेखन, जीनोमिक सूचना जैसे डीएनए अनुक्रम या संरचनाओं के स्थिर पहलुओं के विपरीत जीन प्रतिलेखन, अनुवाद, और प्रोटीन-इंटरैक्शन जैसे गतिशील पहलुओं पर केंद्रित है। कार्यात्मक जीनोमिक्स जीन, आरएनए टेप और प्रोटीन उत्पादों के स्तर पर डीएनए के कार्य के बारे में सवालों के जवाब देने का प्रयास करता है। कार्यात्मक जीनोमिक्स अध्ययनों की एक प्रमुख विशेषता इन सवालों के लिए उनका जीनोम-चौड़ा दृष्टिकोण है, जिसमें आमतौर पर अधिक पारंपरिक "जीन-बाय-जीन" दृष्टिकोण के बजाय उच्च-थ्रूपुट विधियों को शामिल किया जाता है।
जीनोमिक्स की एक प्रमुख शाखा है। अभी भी विभिन्न जीवों के जीनोम अनुक्रमण के साथ संबंध है, लेकिन पूर्ण जीनोम के ज्ञान ने कार्यात्मक जीनोमिक्स के क्षेत्र के लिए संभावना पैदा की है, जो मुख्य रूप से विभिन्न स्थितियों के दौरान जीन अभिव्यक्ति के पैटर्न से संबंधित है। यहां सबसे महत्वपूर्ण उपकरण माइक्रोएरे और जैव सूचना विज्ञान हैं।
संरचनात्मक जीनोमिक्स
संरचनात्मक जीनोमिक्स किसी दिए गए गुंबद द्वारा एन्कोड किए गए प्रत्येक प्रोटीन की 3-आयामी संरचना का वर्णन करना चाहते हैं। यह जीनोम-आधारित दृष्टिकोण प्रयोगात्मक और मॉडलिंग दृष्टिकोणों के संयोजन द्वारा संरचना निर्धारण के एक उच्च-थ्रूपुट विधि के लिए अनुमति देता है। संरचनात्मक जीनोमिक्स और पारंपरिक संरचनात्मक भविष्यवाणी के बीच मुख्य अंतर यह है कि संरचनात्मक जीनोमिक्स एक विशेष प्रोटीन पर ध्यान केंद्रित करने के बजाय जीनोम द्वारा एन्कोड किए गए प्रत्येक प्रोटीन की संरचना को निर्धारित करने का प्रयास करता है। पूर्ण-जीनोम अनुक्रम उपलब्ध होने के साथ, संरचना की भविष्यवाणी प्रायोगिक और मॉडलिंग दृष्टिकोणों के संयोजन के माध्यम से और अधिक तेज़ी से की जा सकती है, खासकर क्योंकि अनुक्रमित जीनोम और पहले से हल किए गए प्रोटीन संरचनाओं की बड़ी संख्या की उपलब्धता वैज्ञानिकों को पहले से हल की संरचनाओं पर प्रोटीन संरचना की अनुमति देती है। homologs। स्ट्रक्चरल जीनोमिक्स में संरचना निर्धारण के लिए बड़ी संख्या में दृष्टिकोण शामिल हैं, जिसमें अनुक्रम या जीनोमिक अनुक्रमों के आधार पर मॉडलिंग-आधारित दृष्टिकोण या संरचनात्मक संरचना के प्रोटीन के लिए संरचनात्मक तरीकों का उपयोग करना शामिल है या एक प्रोटीन के लिए रासायनिक और भौतिक सिद्धांतों पर आधारित है, जिसमें कोई होमोलॉजी नहीं है। किसी भी ज्ञात संरचना। पारंपरिक संरचनात्मक जीव विज्ञान के विपरीत, प्रोटीन जीनोमिक्स के एक संरचनात्मक प्रयास के माध्यम से प्रोटीन संरचना का निर्धारण अक्सर (लेकिन हमेशा नहीं) प्रोटीन फ़ंक्शन के बारे में कुछ भी ज्ञात होने से पहले होता है। यह संरचनात्मक जैव सूचना विज्ञान में नई चुनौतियों को उठाता है, अर्थात इसकी 3 डी संरचना से प्रोटीन फ़ंक्शन निर्धारित करता है।
एपिग्नोमिक्स
एपिगेनॉमिक्स एक कोशिका के आनुवंशिक सामग्री पर एपिगेनियन संशोधनों के पूर्ण सेट का अध्ययन है। एपिजेनोम के रूप में जाना जाता है। एपिजेनेटिक संशोधन एक सेल के डीएनए या हिस्टोन पर प्रतिवर्ती संशोधन होते हैं जो डीएनए अनुक्रम में बदलाव किए बिना जीन अभिव्यक्ति को प्रभावित करते हैं (रसेल 2010 पी। 475)। सबसे विशेषता एपिगेनेटिक संशोधनों में से दो डीएनए मेथिलिकरण और हिस्टोन संशोधन हैं। एपिजेनेटिक संशोधन जीन अभिव्यक्ति और नियमन में एक महत्वपूर्ण भूमिका निभाते हैं, और कई सेलुलर प्रक्रियाओं में शामिल होते हैं जैसे कि भेदभाव / विकास और ट्यूमरजेनिसिस। वैश्विक स्तर पर एपिजेनेटिक्स के अध्ययन को हाल ही में जीनोमिक हाई-थ्रूपुट assays के अनुकूलन के माध्यम से संभव बनाया गया है।
Metagenomics
Metagenomics metagenomes , पर्यावरणीय नमूनों से सीधे आनुवंशिक सामग्री बरामद की गई। व्यापक क्षेत्र को पर्यावरण जीनोमिक्स, इकोजीनॉमिक्स या सामुदायिक जीनोमिक्स के रूप में भी जाना जा सकता है। जबकि पारंपरिक माइक्रोबायोलॉजी और माइक्रोबियल जीनोम अनुक्रमण एक प्राकृतिक नमूने में विविधता की एक प्रोफ़ाइल का उत्पादन करने के लिए क्लोनल संस्कृतियों, प्रारंभिक पर्यावरण जीन अनुक्रमण क्लोनिंग विशिष्ट जीन (अक्सर 16S rRNA जीन) पर भरोसा करते हैं। इस तरह के काम से पता चला कि खेती आधारित तरीकों से अधिकांश सूक्ष्मजीव जैव विविधता छूट गई थी। हाल के अध्ययनों में "शॉटगन" सेंगर अनुक्रमण या बड़े पैमाने पर समानांतर पाइरोइडेंसिंग का उपयोग किया गया है, जो नमूना समुदायों के सभी सदस्यों से सभी जीनों के बड़े पैमाने पर निष्पक्ष नमूने प्राप्त करने के लिए है। सूक्ष्म जीवन की पहले से छिपी विविधता को प्रकट करने की अपनी शक्ति के कारण, मेटागोनॉमिक्स माइक्रोबियल दुनिया को देखने के लिए एक शक्तिशाली लेंस प्रदान करता है जिसमें संपूर्ण जीवित दुनिया की समझ में क्रांति लाने की क्षमता है।
मॉडल सिस्टम बैक्टीरियोफेज ने जीवाणु आनुवंशिकी और आणविक जीव विज्ञान में महत्वपूर्ण भूमिका निभाई है और जारी है। ऐतिहासिक रूप से, उनका उपयोग जीन संरचना और जीन विनियमन को परिभाषित करने के लिए किया गया था। इसके अलावा अनुक्रमित होने वाला पहला जीन एक बैक्टीरियोफेज था। हालांकि, बैक्टीरियोफेज अनुसंधान ने जीनोमिक्स क्रांति का नेतृत्व नहीं किया, जो स्पष्ट रूप से बैक्टीरिया जीनोमिक्स द्वारा हावी है। केवल बहुत हाल ही में बैक्टीरियोफेज जीनोम का अध्ययन प्रमुख हो गया है, जिससे शोधकर्ताओं को चरणबद्ध विकास अंतर्निहित तंत्र को समझने में सक्षम बनाया गया है। बैक्टीरियोफेज जीनोम अनुक्रम को अलग-अलग बैक्टीरियोफेज के प्रत्यक्ष अनुक्रमण के माध्यम से प्राप्त किया जा सकता है, लेकिन इसे माइक्रोबियल जीनोम के हिस्से के रूप में भी प्राप्त किया जा सकता है। बैक्टीरियल जीनोम के विश्लेषण से पता चला है कि पर्याप्त मात्रा में माइक्रोबियल डीएनए में प्रोफ़ेज अनुक्रम और प्रोफ़ेज जैसे तत्व होते हैं। इन अनुक्रमों का एक विस्तृत डेटाबेस खनन बैक्टीरिया जीनोम को आकार देने में भविष्यवाणियों की भूमिका में अंतर्दृष्टि प्रदान करता है: कुल मिलाकर, इस विधि ने कई ज्ञात बैक्टीरियोफेज समूहों को सत्यापित किया, जिससे यह बैक्टीरियल मुहावरों से भविष्यवाणियों के संबंधों की भविष्यवाणी करने के लिए एक उपयोगी उपकरण है।
वर्तमान में 24 साइनोबैक्टीरिया हैं जिनके लिए कुल जीनोम अनुक्रम उपलब्ध है। इनमें से 15 सायनोबैक्टीरिया समुद्री वातावरण से आते हैं। ये छह प्रोक्लोरोकोकस उपभेदों, सात समुद्री Synechococcus उपभेदों, ट्राइकोडेसियम एरिथ्रियम IMS101 और Crocosphaera watsonii WH8501 हैं। कई अध्ययनों से पता चला है कि समुद्री साइनोबैक्टीरिया के महत्वपूर्ण पारिस्थितिक और शारीरिक विशेषताओं का पता लगाने के लिए इन अनुक्रमों का बहुत सफलतापूर्वक उपयोग कैसे किया जा सकता है। हालांकि, वर्तमान में कई और जीनोम परियोजनाएं चल रही हैं, उनमें से आगे हैं प्रोक्लोरोकोकस और समुद्री सिन्टोकॉकस अलग-थलग, एक्रायोक्लोरिस / / i> और प्रोक्लोरन , एन 2-फिक्सिंग फिलामेंटस सियानोबैक्टीरिया नोडुलरिया स्पुमीगेना , लिंग्बुइ सौजुरी और लिंगबाग माजुस्कुल्टा , साथ ही समुद्री सियानोबैजेरिया को संक्रमित करने वाले जीवाणु। इस प्रकार, जीनोम जानकारी के बढ़ते शरीर को तुलनात्मक दृष्टिकोण लागू करके वैश्विक समस्याओं को संबोधित करने के लिए और अधिक सामान्य तरीके से टैप किया जा सकता है। इस क्षेत्र में प्रगति के कुछ नए और रोमांचक उदाहरण नियामक आरएनए के लिए जीन की पहचान हैं, प्रकाश संश्लेषण के विकास मूल में अंतर्दृष्टि, या विश्लेषण किए गए जीनों को क्षैतिज जीन हस्तांतरण के योगदान का अनुमान है।
जीनोमिक्स के अनुप्रयोग
जीनोमिक्स ने चिकित्सा, जैव प्रौद्योगिकी, नृविज्ञान और अन्य सामाजिक विज्ञानों सहित कई क्षेत्रों में आवेदन प्रदान किए हैं।
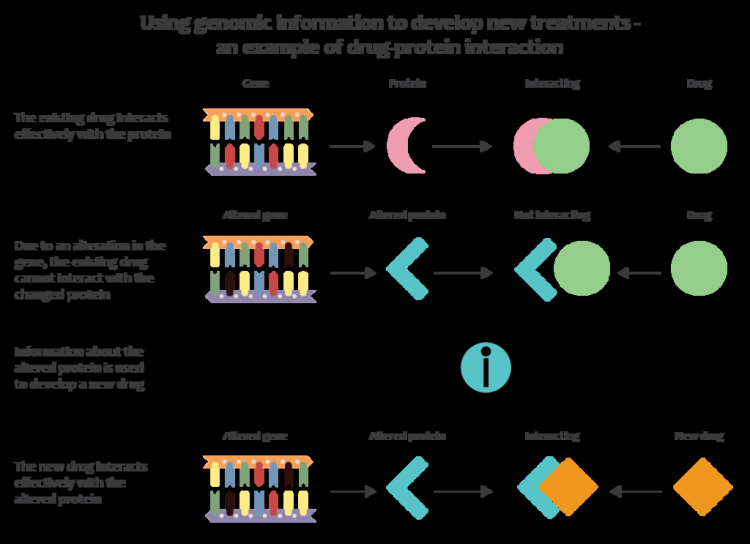
जीनोमिक दवा
अगली पीढ़ी के जीनोमिक। प्रौद्योगिकियां चिकित्सकों और बायोमेडिकल शोधकर्ताओं को बड़ी अध्ययन आबादी पर एकत्र जीनोमिक डेटा की मात्रा में काफी वृद्धि करने की अनुमति देती हैं। जब नए सूचना विज्ञान के साथ संयुक्त किया जाता है जो रोग अनुसंधान में जीनोमिक डेटा के साथ कई प्रकार के डेटा को एकीकृत करता है, तो यह शोधकर्ताओं को दवा प्रतिक्रिया और रोग के आनुवंशिक आधारों को बेहतर ढंग से समझने की अनुमति देता है। जीनोम को दवा के लिए लागू करने के शुरुआती प्रयासों में उन लोगों में शामिल थे, जिन्हें स्टैनफोर्ड की टीम ने यूआन एशले के नेतृत्व में शामिल किया, जिन्होंने मानव जीनोम की चिकित्सा व्याख्या के लिए पहले उपकरण विकसित किए। उदाहरण के लिए, हमारे सभी अनुसंधान कार्यक्रम का उद्देश्य 1 मिलियन प्रतिभागियों से जीनोम अनुक्रम डेटा एकत्र करना है, जो सटीक चिकित्सा अनुसंधान मंच का एक महत्वपूर्ण घटक बन जाता है।
सिंथेटिक जीव विज्ञान और जैव अभियांत्रिकी <। / h3>
जीनोमिक ज्ञान की वृद्धि ने सिंथेटिक जीव विज्ञान के तेजी से परिष्कृत अनुप्रयोगों को सक्षम किया है। 2010 में जे। क्रेग वेंटर इंस्टीट्यूट के शोधकर्ताओं ने माइकोप्लास्मा जननांग
के जीनोम से व्युत्पन्न जीवाणु की एक आंशिक रूप से सिंथेटिक प्रजाति, मायकोप्लाज्मा लेबरटोरियम के निर्माण की घोषणा की।संरक्षण जीनोमिक्स
जीनोमिक अनुक्रमण द्वारा एकत्र की गई जानकारी का उपयोग प्रजातियों के संरक्षण के लिए आनुवंशिक कारकों की बेहतर मूल्यांकन करने के लिए किया जा सकता है, जैसे कि जनसंख्या की आनुवंशिक विविधता या एक व्यक्ति एक पुनरावर्ती के लिए विषमलैंगिक है या नहीं। आनुवंशिक विकार विरासत में मिला। विकासवादी प्रक्रियाओं के प्रभावों का मूल्यांकन करने के लिए जीनोमिक डेटा का उपयोग करके और किसी दिए गए जनसंख्या में भिन्नता के पैटर्न का पता लगाने के लिए, संरक्षणवादी किसी दिए गए प्रजाति की सहायता के लिए योजना बना सकते हैं, क्योंकि बिना मानक चर दृष्टिकोण के अनजाने में कई चर अज्ञात हैं।
Gugi Health: Improve your health, one day at a time!

जीने के लिए स्वस्थ तरीके से कम
स्वास्थ्य पत्रिका से आप काम करते हैं, आप सही खाने की कोशिश करते हैं, और आप …

जीन्स भी खर्राटे? क्यों तुम बाहर बेकार नहीं होना चाहिए
"कोई भी वापस नहीं जा सकता है और एक नई शुरुआत कर सकता है, लेकिन कोई भी आज शुरू कर …
